

Research Activities
Theory and simulation of surface Sum Frequency Generation (SFG) spectroscopy
Molecular modeling of electronic polarization
--- effects on molecular dynamics in solutions ---
Mass transfer kinetics in heterogeneous atmospheric chemistry
Free energy calculations for chemical reactions in condensed phase
Our research activities focus on various molecular processes in solutions and interfaces. Our interests are mainly in the processes not properly described by current theory or experimental interpretation. Development of original theoretical avenues is therefore most emphasized in our group, as illustrated below.
Our theoretical backgrounds are quantum chemistry of molecular electronic structure and molecular dynamics simulation for condensed phase structure and dynamics. Sometime computational fluid dynamics or statistical theories of solutions are employed. Massive computations are carried out using the advanced IMS computer facilities.
Theory and simulation of surface Sum Frequency Generation (SFG) spectroscopy
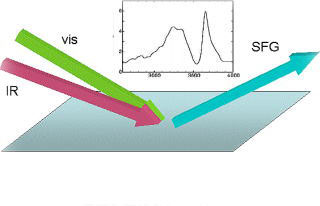
Infrared-visible Sum Frequency Generation (SFG) spectroscopy has wide applicability to interface characterization, including wet surfaces in ambient conditions and buried interfaces such as liquid-liquid or liquid-solid interfaces. It is also suitable to dynamics study of surface molecules, by exploiting the time resolution of the laser pulses.
In contrast to the recent experimental progress, however, theoretical aspects of SFG are still far from complete. We recently proposed two theories of SFG, which enable us to compute SFG spectra via quantum chemistry and molecular dynamics calculations for the first time.
This project aims at extending and refining our SFG theories toward general and practical use for experimental interpretation. We have sub-projects in this category; (1) development of fundamental SFG theory toward quantitative interpretation of the experiments, and (2) application to many interfacial structure of interest using massive computation.
Molecular modeling of electronic polarization
--- effects on molecular dynamics in solutions ---
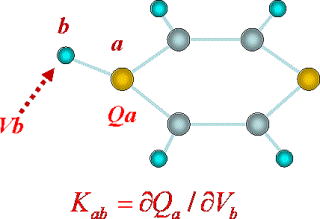
In molecular modeling of interaction potential, the short-range repulsive part mainly determines the shape of a molecule, whereas the electrostatic interaction shows particularly large variety, reflecting chemical characters of molecular electronic structure. Electronic polarization is a substantial part of the latter, and it often plays critical roles in condensed phase dynamics. In actual solutions, the electronic polarization fluctuates with molecular motions.
We have developed a new theory of electronic polarization, namely the charge response kernel (CRK), on the basis of ab initio molecular orbital theory. Since the CRK is free from empirical parameterization, it provides a general description of intramolecular charge fluctuation of any molecules, including reactive intermediates or unstable species. We applied this theory to intermediate aromatic radicals or azide ion, and succeeded to elucidate anomalous diffusion of aromatic radicals or fast vibrational relaxation of azide ion (N3-), which were experimental but puzzling facts, by implementing the theory in molecular dynamics simulation.
The potential of this CRK theory has not been fully exploited yet. It is also quite useful to the sum frequency generation theory described above, and we are now implementing the CRK theory in the SFG theory.
In order to precisely model the electronic polarization in solutions, electronic polarization should be treated in the condensed phase, not in the gas phase. We have demonstrated that a molecular polarizability is in fact perturbed by the surrounding solvent. One substantial mechanism is that the solution-solvent exchange repulsion confines the spatial tail of the electronic cloud.
Mass transfer kinetics in heterogeneous atmospheric chemistry
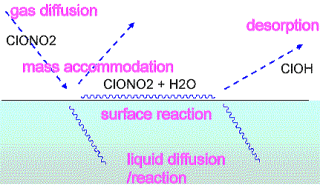
In the modern atmospheric science, heterogeneous processes involving aerosols and clouds are drawing particular attention, in connection to stratospheric ozone destruction, acid rain, and direct/indirect effects on global warming. Despite of their importance, these processes are quite challenging to molecular science. Much work is needed to molecular-level understanding of them, including very basis issues of heterogeneous phenomena.
One fundamental problem is mass transfer kinetics at liquid-vapor interfaces. Experimentally observed kinetics often range over several orders of magnitude and disagree each other. It is a very difficult subject to interpret in a microscopic sense, since phenomenological kinetics includes many factors associated to gas, liquid and surface. Theoretical analysis of the heterogeneous kinetics is greatly needed, and we have demonstrated that computational fluid dynamics and molecular dynamics calculations are in fact very powerful to decompose into elemental kinetic factors.
In order to understand kinetics and dynamics at aerosol surfaces, information on the aerosol surface structure is necessary. The SFG spectroscopy is a particularly promising tool for the surface characterization in atmospheric conditions.
Free energy calculations for chemical reactions in condensed phase
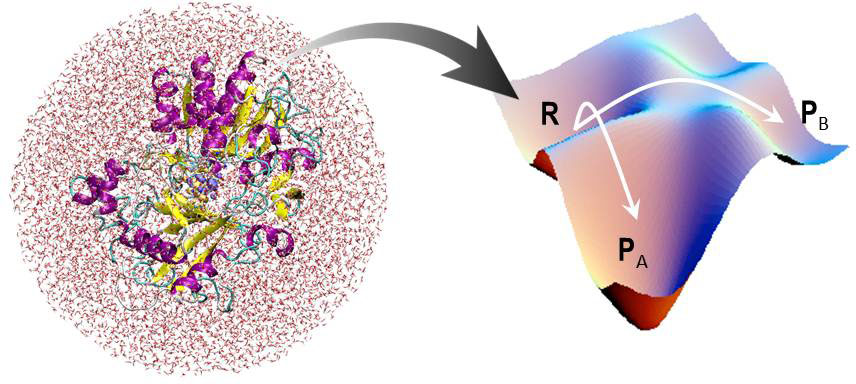
To understand chemical reaction mechanisms in condensed phase, information on the free energy surface is of vital importance. However, evaluating the free energy surfaces for realistic chemical reactions is a crucial but challenging issue to computational molecular science, since we have to take account of both the electronic state of reacting species and thermal fluctuation of surrouding solvents. In order to overcome these difficulties, we have developed a novel method of QM/MM calculation in combination of integral equation theory for condensed systems. This method realizes computationally efficient and accurate evaluation of the free energies beyond the conventional QM/MM calculations.
Applications of this free energy calculation method cover wide range of chemical reactions in condensed phase, including isomerization, acid/base reactions, enzyme reactions, and heterogeneous reactions at interfaces. This method is particularly suitable to the reactions in heterogeneous environment of solvation, such as interfaces or nano-confined space, which are often beyond the applicability of conventional continuum modeling of solvation.