

| 研究テーマ | 教授のメッセージ | 輪読資料 |
研究テーマ
溶液内や界面での分子過程を、電子状態計算や分子シミュレーションに基づく理論計算によって解明する研究を展開しています。 最近では溶液中の化学反応やダイナミックスに加えて、不均質系の界面現象を主な研究対象としています。
凝集系での化学現象を分子レベルで理解するには、個々の分子の化学的な特徴と分子間力が織りなす統計的な性質の両方が必要です。 私たちの研究グループでは電子状態と分子動力学の両方の知識をふまえて、新しい理論的方法論の開発や応用を図っています。 以下で研究課題に挙げた界面和周波発生の理論や自由エネルギー計算の理論などは、その典型的な例です。 また新しい計算手法を、スーパーコンピュータを最大限に用いた大規模計算の中に実現していきます。
電子状態に基づく分子モデリングと分子動力学シミュレーションを主な方法とする理論・計算化学は、凝集系を解明する上で応用範囲の広い正攻法といえ、近年の計算機の進歩に伴って、化学の中での重要性を急速に増しつつあります。
計算化学の将来を担う学生さんには、実験と協力できる幅広い視野と理論計算の研究手法を身に付け、実験だけでは解明できない領域に答えを与えられる研究を目指してほしいと思っています。
界面和周波発生分光の理論とシミュレーション
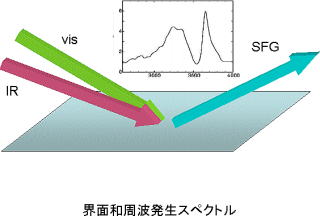
界面を分子レベルで構造解析できる手法として、赤外−可視光の組み合わせによる和周波発生(SFG)分光が、近年では広く用いられるようになってきました。 この方法は、液体・高分子表面や大気圧下での表面、液-液、固-液界面のような埋もれた界面など、従来の表面科学で扱うことの難しかった未開拓の界面現象への応用が特に期待されています。 また、パルスレーザーの時間分解を生かした表面吸着分子のダイナミックスの研究にも適しています。
しかし近年の実験の急速な進展と普及に比べて、理論面は立ち後れていました。 和周波発生の一般論はあるのですが、物質に即して実験データを解析できるレベルには達していませんでした。 私たちは電子状態理論と分子動力学シミュレーションに基づいて、SFGスペクトルを非経験的に計算し解析する理論を初めて提唱し、大きな注目を受けました。 SFG分光から界面の情報を正確に引き出すには、理論計算が非常に有効であることを明らかとしてきました。 しかし今後この理論を真に実用化し、実験と理論計算の広い協力体制を確立するためには、これから多くの仕事が必要です。
現在私たちは、(1)界面SFGの基礎理論の発展・深化、および(2)分子モデルの開発と大規模計算への応用を進めています。 水溶液界面や有機薄膜、固液界面での吸着構造など興味ある系に応用するには大規模計算が必要で、分子研の計算資源などもフルに活用していきます。 この研究は、現在の森田研究室の主力テーマの一つとなっています。
電子分極の理論と分子モデリングへの応用
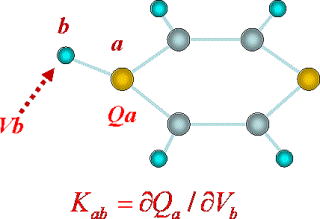
溶液内での分子のダイナミックスを理解する上で、電子分極の効果はしばしば非常に重要です。 分子間力のなかで分子の化学的個性が特に顕著に現れる部分で、実際の溶液中では分子の運動とともに電子分極もゆらいでいます。
そこで分子内の電子分極を表す理論として、通常の分極率よりも分子サイトの性質を反映する量 (Charge Response Kernel) を、分子軌道法を用いて定式化しました。 この理論は完全にab initio法に基づくために経験的な物性量をモデル化に必要とせず、反応中間体などの不安定種や新奇な分子なども共通して扱うことができます。 それをもとに芳香族ラジカルの拡散速度やアジドイオンの振動寿命など、従来説明がつかないとされていた溶液内ダイナミックスの実験事実を、電子分極の効果として解明しました。
この理論は、電子分極を含んだ分子モデルを一般的に与えることが可能で、分子軌道法や密度汎関数法に基づく理論開発やプログラミングも行われてきました。 特に上の界面和周波発生の計算には非常に有効で、現在その方向への展開を進めています。 またこの電子分極の理論の発展として、溶液の統計力学理論との連携も考えられ、 以下に示すQM/MM法の中に取り入れて融合する研究も現在進んでいます。
溶液中での分子の分極率はしばしば気相分子の値として扱われますが、それはどこまで正しいでしょうか。 私たちは電子状態計算によって、水中での溶媒和が分極率に無視できない影響を与えることを示しました。 これは主として溶質−溶媒分子の電子間交換反発によるもので、この効果を正しく評価することは分子モデルの定量化にとって残された問題です。
液体界面での物質移動と不均質大気化学反応
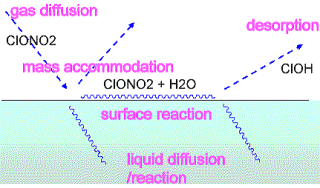
現在の大気環境化学では、気相均一系のみならず、エアロゾルとよばれる微粒子が関与する不均質化学が極めてホットな問題となっています。 これは成層圏オゾン破壊、酸性雨、地球温暖化への直接・間接(雲形成を介した)効果など現代的な問題にとって、非常に重要かつ理解が遅れた分野であるためです。 しかし物理化学としては未開拓で、今後のターゲットとなるべき問題が多く残されています。
その中の基礎的な問題の一つとして、気液界面での物質移動があり、これは従来何桁にもわたる甚だしい曖昧さがしばしば報告されていました。 実験的に測定される界面取り込み速度は純粋な界面現象ではなく、気相拡散や液相での溶解度など多くの要素を含んだ複雑なもので、実験でそれらの要素を曖昧さなく分離することは容易ではありません。 私たちは流体計算と分子動力学計算を用いて、実験で現れる要素ステップを実際に分離して解析できることを初めて示しました。 その結果、液体界面の物質移動ダイナミックスについて実験と理論計算を精密に比較して研究する道が開かれてきました。
液体界面での物質移動は、多くの分野に関係する問題です。 エアロゾル表面での化学反応のみならず、気液二相流のなかでの気泡の安定性、水‐油の液液界面での抽出や相間移動触媒など、分子レベルの理解が求められる多くの液体界面の関わる問題を今後解明していきます。
溶液中での化学反応過程と自由エネルギー計算
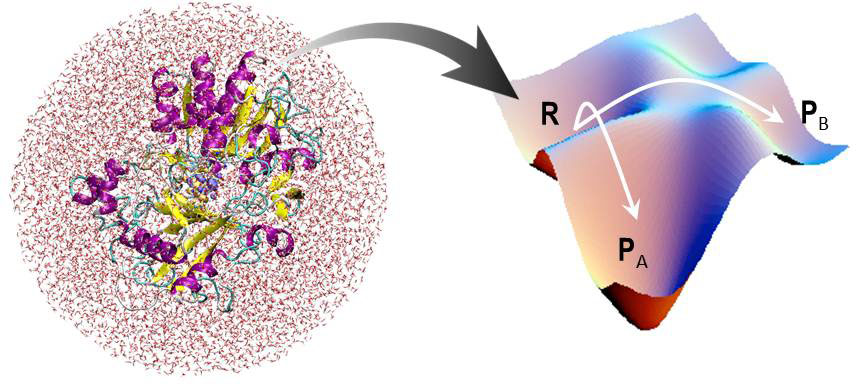
化学反応を記述する最も正攻法は、化学反応経路に沿った自由エネルギー面を理解することといえます。 しかし一般に凝縮相での自由エネルギーを実際の化学反応に即して求めることは、計算分子科学としてチャレンジングな課題です。 化学結合や電子状態の変化を伴う過程を表すには量子化学が必要であり、かつ周囲の溶媒が熱ゆらぎをしている効果も重要であるためです。 そこで我々は密度汎関数法に基づく量子化学と分子力場を組み合わせた手法(QM/MM法)と溶液の分子シミュレーションや積分方程式理論を併用して、溶液中での自由エネルギーを効率良く求める一般的で強力な手法を開発してきました。
この手法の応用範囲は極めて広く、溶液中での異性化反応や酸塩基反応などから生体中での酵素反応、界面での不均質反応まで、多くの化学反応が研究の対象となっています。 とくに界面やナノ空間など、従来の誘電体モデル等で扱うことが難しい複雑な環境への応用に適しており、凝集系での化学反応の解明を広げる役割が期待されています。